


Universitätsklinikum Essen
Ihr führendes Gesundheits- und Kompetenzzentrum in der Metropolregion Ruhrgebiet
Willkommen auf der Website des Universitätsklinikums Essen
Ihr top ausgezeichnetes Klinikum im Herzen des Ruhrgebiets.
Das Universitätsklinikum Essen ist Teil des Klinikverbunds Universitätsmedizin Essen. Zu diesem gehören 15 weitere Tochterunternehmen, unter anderem die Ruhrlandklinik, das St. Josef Krankenhaus Werden sowie die Herzchirurgie Huttrop und das Westdeutsche Protonentherapiezentrum Essen.
Die Essener Universitätsmedizin ist mit etwa 11.000 Beschäftigten und 1.700 Betten das führende Gesundheits-Kompetenzzentrum des Ruhrgebiets und seit 2015 auf dem Weg zum Smart Hospital.
Das Universitätsklinikum Essen setzt seit Jahren erfolgreich auf die Schwerpunkte Onkologie, Transplantation, Herz-Kreislauf, Infektiologie/Immunologie, sowie Translationale Neuro- und Verhaltenswissenschaften.
Einrichtungen



Zahlen der Dachgesellschaft Universitätsmedizin Essen
Kliniken
Institute
Beschäftigte
Aktuelle Corona-Maßnahmen
Aufgrund der derzeitigen Covid-19-Situation sind wir gezwungen, unsere verschärften Hygienemaßnahmen aufrecht zu halten. Die aktuellen geltenden Regelungen je nach Standort des UME Klinikverbundes können Sie auf unserer Seite nachlesen.

Wir geben neuen Pflegekräften eine Perspektive
Die Universitätsmedizin hat 150 neue Stellen in der Krankenpflege geschaffen.
Werden Sie Teil unseres Teams in einer unserer 33 Kliniken!
Social Media
Spenden Sie lebenswichtiges Blut
Und helfen Sie Menschen in Not mit einer Blutspende bei der Transfusionsmedizin des Universitätsklinikums Essen.

Patienten + Besucher
Institut für PatientenErleben
Unter PatientenErleben verstehen wir an der Universitätsmedizin Essen alle Erfahrungen, die unsere Patientinnen und Patienten rund um den Klinikaufenthalt machen. Hierzu zählt alles, was sie sehen, hören, berühren und denken. Diese Eindrücke, die bewusst oder unbewusst sein können, bilden das individuelle PatientenErleben.


Unterstützen Sie den Bau der neuen Kinderklinik
Eine Klinik für die Kleinen, in der sie das Kind sein nicht vergessen müssen – mit Ihrer Spende und unserer neuen Kinderklinik machen wir es gemeinsam möglich.
Für Ärzte


Ukraine-Support der Universitätsmedizin Essen
Erfahren Sie mehr über aktuelle Spendenaktionen der Stiftung Universitätsmedizin.

Westdeutsches Zentrum für Organtransplantation
Wer ein Spenderorgan benötigt, ist in den meisten Fällen schwer krank. Um betroffene Patientinnen und Patienten auf medizinisch höchstem Niveau zu behandeln, erforscht das ärztliche Team und Wissenschaftler und Wissenschaftlerinnen der Universitätsmedizin Essen seit vielen Jahrzehnten neue Erkenntnisse und Verfahren. Im Westdeutschen Zentrum für Organtransplantation zusammengeschlossen, bündeln sie dort ihre Expertise zum Wohle von Patienten und Patientinnen.

Der Organspendeausweis
Informieren Sie sich jetzt rund um das Thema Organspendeausweis.

Forschung + Lehre
Die Forschung in der Medizinischen Fakultät der Universität Duisburg-Essen am Universitätsklinikum Essen konzentriert sich auf die Schwerpunkte Herz-Kreislauf, Onkologie, Transplantation
sowie Translationale Neuro- und Verhaltenswissenschaften, Infektiologie und Immunologie.
Die Ansiedelung von Fakultät und Universitätsklinikum auf einem Campus ermöglicht dabei eine enge Verzahnung von Forschung und klinischer Versorgung, durch die wir neue Erkenntnisse schnell in der Patientenversorgung nutzbar machen können.

Beruf + Familie
Erhalten Sie Unterstützung bei allen Fragen zur Vereinbarkeit von Beruf und Familie – in unserem MitarbeiterServiceBüro.
Beruf + Karriere

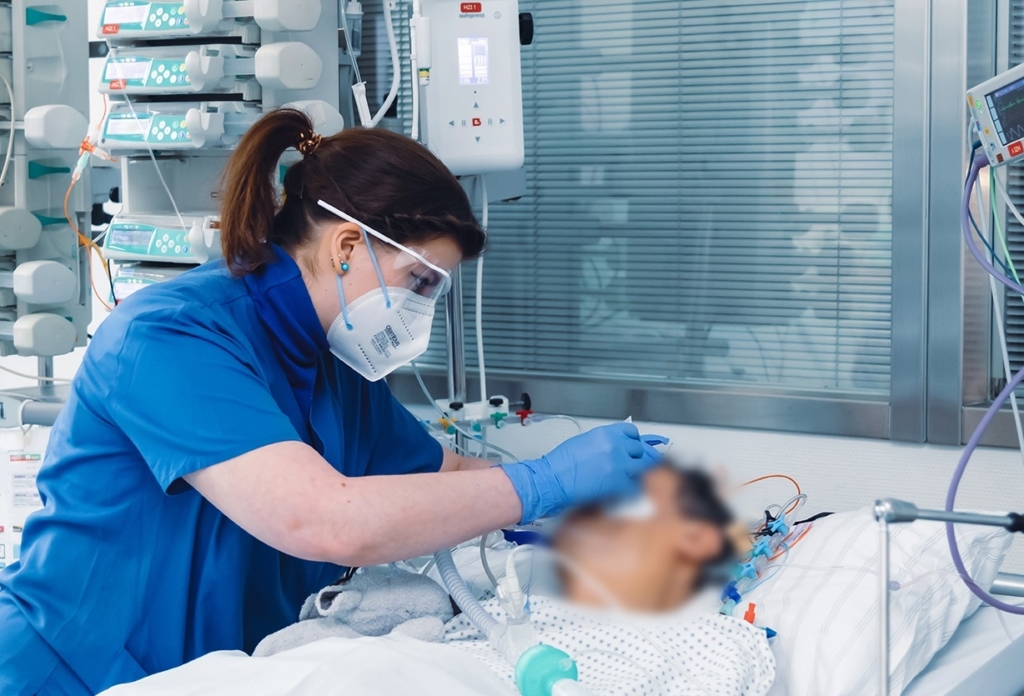
Jetzt Chance ergreifen!
Von der Medizin und Pflege bis hin zu den zahlreichen Mitarbeitenden in Küche, Transport und Technik – das Universitätsklinikum Essen ist mit seinen rund 8.000 Beschäftigten einer der führenden Arbeitgeber im Ruhrgebiet.
Darüber hinaus bilden wir zurzeit rund 700 junge Menschen in unterschiedlichen Bereichen aus. Darunter die Gesundheits- und Krankenpflege, Physiotherapie sowie auch technische Berufe.
In unserer Jobbörse finden Sie Stellenangebote für wissenschaftliche sowie nicht wissenschaftliche Beschäftigte, Pflegeberufe sowie Ausbildungs- und Praktikumsplätze. Bitte beachten Sie zusätzlich die Stellenausschreibungen auf den Seiten der jeweiligen Kliniken und Institute.

Respekt vor Ihrem Engagement
Der gesamte Vorstand möchte sich ganz herzlich bei seinen Mitarbeitenden und ihren Familien für ihr beeindruckendes Engagement bedanken.

Über Uns

Jahresbericht 2022
Seit 2021 erscheint unser Jahresbericht mit Blick auf die Nachhaltigkeit digital. Filme, Foto-Reportagen, Interviews, Meldungen und Fakten finden Sie auf unserer Microsite für den Jahresbericht. Schauen Sie doch mal rein!
Nachhaltigkeitsmanagement
Nachhaltig denken und handeln ist in der heutigen Zeit vor allem zum Schutz unserer natürlichen Lebensgrundlage unabdingbar. Nachhaltigkeit betrifft dabei jedoch nicht nur unsere Umwelt, sondern alle Bereiche unseres Lebens und Wirtschaftens. Nachhaltiges Handeln ist also eine Aufgabe der gesamten Gesellschaft – von uns als Privatperson, aber auch im speziellen von uns als Maximalversorger im Gesundheitswesen.
Unsere Mission als Universitätsmedizin Essen ist der Weg zu einem nachhaltigen und grünen Krankenhaus, im Sinne unserer Patienten, Mitarbeiter und schlussendlich für uns alle.

Veranstaltungen
Qualitätsberichte
Lesbare Versionen der strukturierten Qualitätsberichte nach § 136b Abs. 1 Satz 1 Nr. 3 SGB V.

